
July 2024
Outlook on Rare Neurodegenerative Diseases: ALS and SMA
Rare CNS diseases are defined by the specialized care they require; recent technological innovations and commercial incentives have spurred investment in this area.
Rare Central Nervous System Diseases – Overview
Rare CNS diseases are highly morbid and associated with high mortality and debilitating symptoms that require chronic treatment by a neurologist or other specialists (e.g., epileptologists, neuromuscular specialists). Care is often provided at centers of excellence (CoE) that offer comprehensive multi-disciplinary approach to disease management.
(1994-2024)1, 2
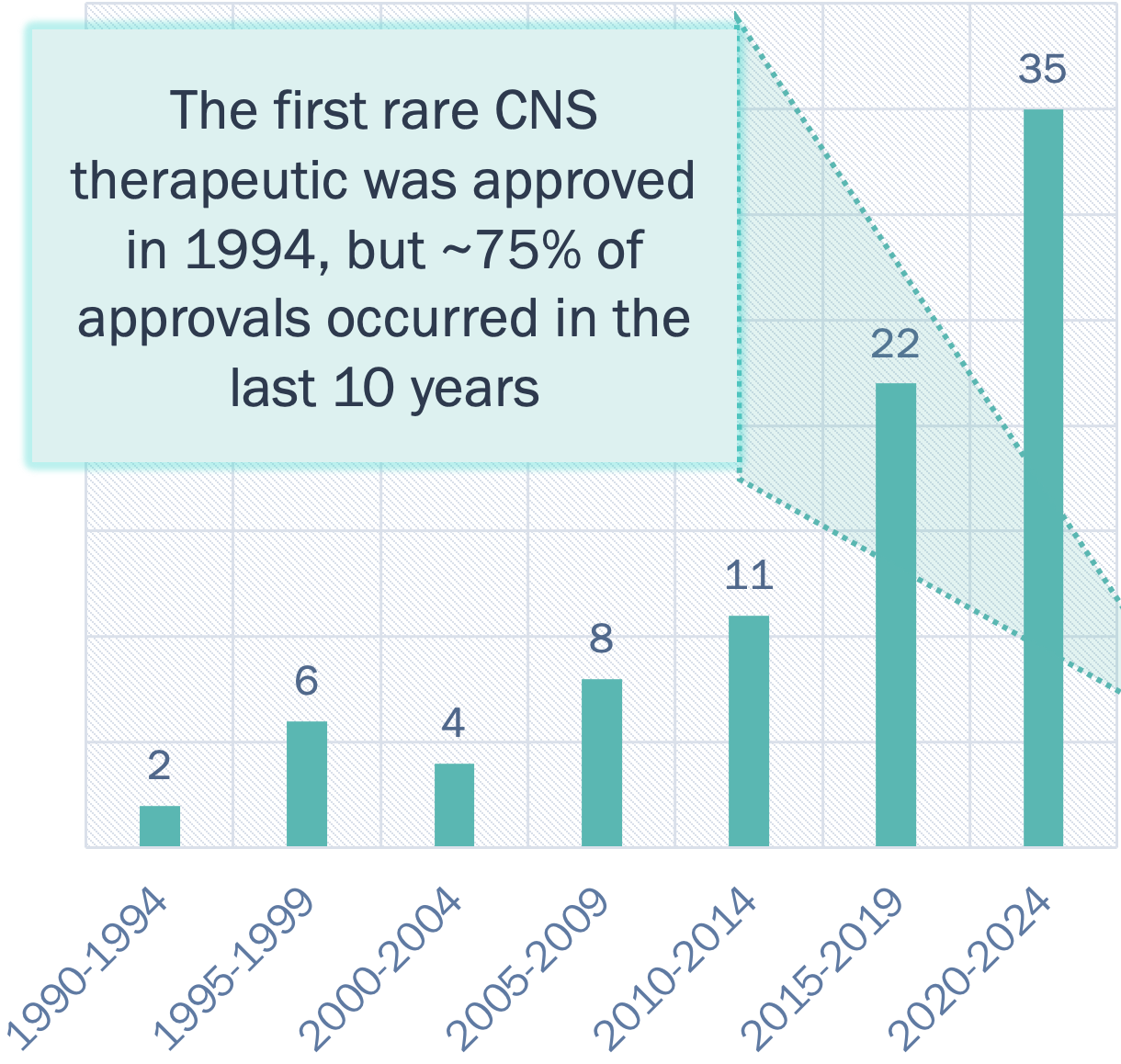
Incentives exist for developing rare disease therapies pre and post approval:
- Expedited drug review: The high disease burden and substantial clinical unmet need often make rare neurodegenerative drugs eligible for fast track or accelerated approval pathways and priority review vouchers.
- Concentrated sales force: Rare diseases are often treated at CoE.
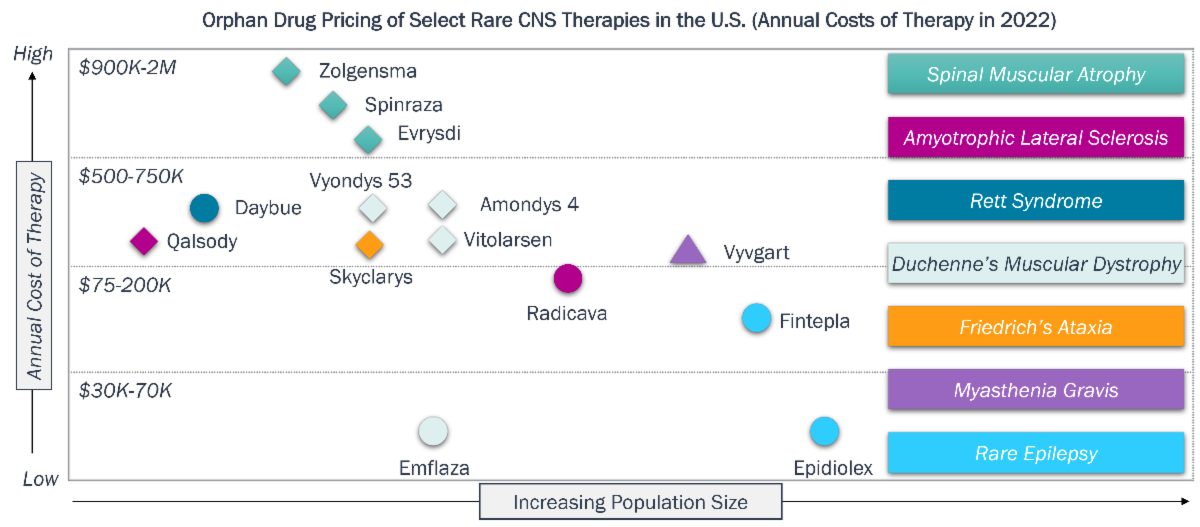
- Orphan drug pricing and exclusivity: Orphan drugs are granted seven-year market exclusivity in the U.S. and 10-year exclusivity in E.U. Orphan drugs, including those for rare neurodegenerative diseases, command significant pricing power in the U.S., with the average cost reported to be $220K/patient/year compared to $13K/patient/year for non-orphan drugs.
Emergence of novel technologies:
- 80% of rare CNS diseases are genetic in origin, making them an attractive target for companies developing gene-based approaches (e.g., gene editing, gene replacement).
- Improved, and less invasive delivery technologies to enhance blood brain barrier penetration or biodistribution are also on the rise (e.g., QurAlis' Flex-ASO).
Source: (1) FDA website (2) Pharmaprojects (3) Kioutchoukova, Ivelina P., et al. "World Journal of Experimental Medicine" 13.4 (2023): 59. (4) Althobaiti, Hana, et al. Healthcare. Vol. 11. No. 4. MDPI, 2023
ALS and SMA are two rare neurodegenerative CNS diseases of differing pathophysiological complexity and available treatment options
Background on ALS and SMA
- ALS is a fatal neurodegenerative disease that affects the brain and spinal cord, which causes gradual deterioration and death of motor neurons that control voluntary muscle movements; the average life expectancy from the time of diagnosis is two to five years, but some patients may live for decades.
- Most cases are sporadic, i.e. present with no family history, while 15% are familial in origin (e.g., SOD1 or FUS mutations).
- Disease is highly heterogenous clinically including region of onset (bulbar or limb), degree of motor neuron involvement, rate of disease progression, and presence of cognitive/behavioral changes.
- Therapies approved for ALS (FDA approval dates) include:
- Riluzole (Rilutek, Tiglutik, Exservan, 1995): believed to reduce motor neuron damage as a glutaminergic blocker
- Nuedexta (2010): indicated specifically for pseudobulbar affect symptoms via sigma-1 agonist/NMDA receptor antagonist activity
- Edaravone (Radicava, 2017): believed to act as a neuroprotective agent that prevents oxidative stress damage
- Tofersen (Qalsody, 2023): antisense oligonucleotide that blocks SOD1 protein production; established neurofilament (NfL) as a biomarker
- Relyvrio (2022): believed to prevent nerve cell death; withdrawn in April 2024 after confirmatory trial failure
- SMA is a neuromuscular disease that consists of degeneration of a subset of nerve cells in the spinal cord and motor nuclei in the brainstem, resulting in progressive muscle weakness and atrophy.
- SMA is genetically driven by biallelic mutations in the survival motor neuron 1 (SMN1) gene; loss of SMN1 may be partially compensated for by a back-up gene SMN2, with more SMN2 copies being associated with less severe phenotypes.
- Traditionally SMA consisted of three subtypes, which have now been expanded to five (type 0 to type 4), which are defined by age of onset and the progression of loss of motor function; type 0 and 1 are the most severe and common forms with a life expectancy of less than two years; type 3 appears around 18 months of age and although life expectancy is similar to the general population, type 3 is characterized by difficulties walking; type 4 appears in adulthood and these individuals remain active and enjoy a normal life expectancy.
- Therapies approved for SMA (FDA approval dates) include:
- Nusinersen (Spinraza, 2016): antisense oligonucleotide (ASO) that modifies splicing of SMN2 to upregulate expression of the compensatory splice form of SMN2
- Onasemnogene abeparvovec-xioi (Zolgensma, 2019): AAV9 based gene therapy that provides a functional copy of SMN1
- Risdiplam (Evrysdi, 2020): small molecule splicing modifier that upregulates the compensatory splice form of SMN2
Multiple incentives offset high clinical development risks and costs for drug modalities of all types, as exemplified by marketed drugs for SMA and ALS in the U.S.
Rare CNS Diseases – Clinical and Commercial Incentives
Legend:
![]() Disease Modifying Mechanism of Action (MoA)
Disease Modifying Mechanism of Action (MoA)
![]() Understood MoA for
Symptomatic Treatments
Understood MoA for
Symptomatic Treatments
![]() Unclear Biological Rationale
Unclear Biological Rationale
| ALS | SMA |
|---|---|
| Clinical Development Incentives | |
| Radicava and Qalsody were granted priority review and fast track designations; Qalsody was also approved via accelerated pathway. | Zolgensma was granted breakthrough designation; Spinraza and Everysdi were granted pediatric priority review vouchers. |
| Specialized Care Means Lower Marketing Spend | |
| ALS specialists at CoEs are available to patients, e.g. centers certified by ALS Association. | SMA patients are identified by newborn screening (NBS) and referred to specialized centers via networks such as CureSMA. |
| Commercial Performance (Select Examples) | |
| Radicava, approved in 2017, generated $224M in revenues in 2023 (U.S), and ~15K patients in the U.S. have been treated. | Evrysidi, approved in 2020, generated $621M in revenues in 2023 (U.S.), and ~8.5K patients have been treated globally. |
Rare CNS diseases vary in their ‘druggability’ and unmet need; ALS and SMA are on opposite ends of this spectrum resulting in distinct drug development and clinical trial strategies
Rare CNS Diseases – Druggability and Unmet Need
Spectrum of Rare CNS Diseases – Select
Examples
(Level of Unmet Need vs. Disease Complexity)
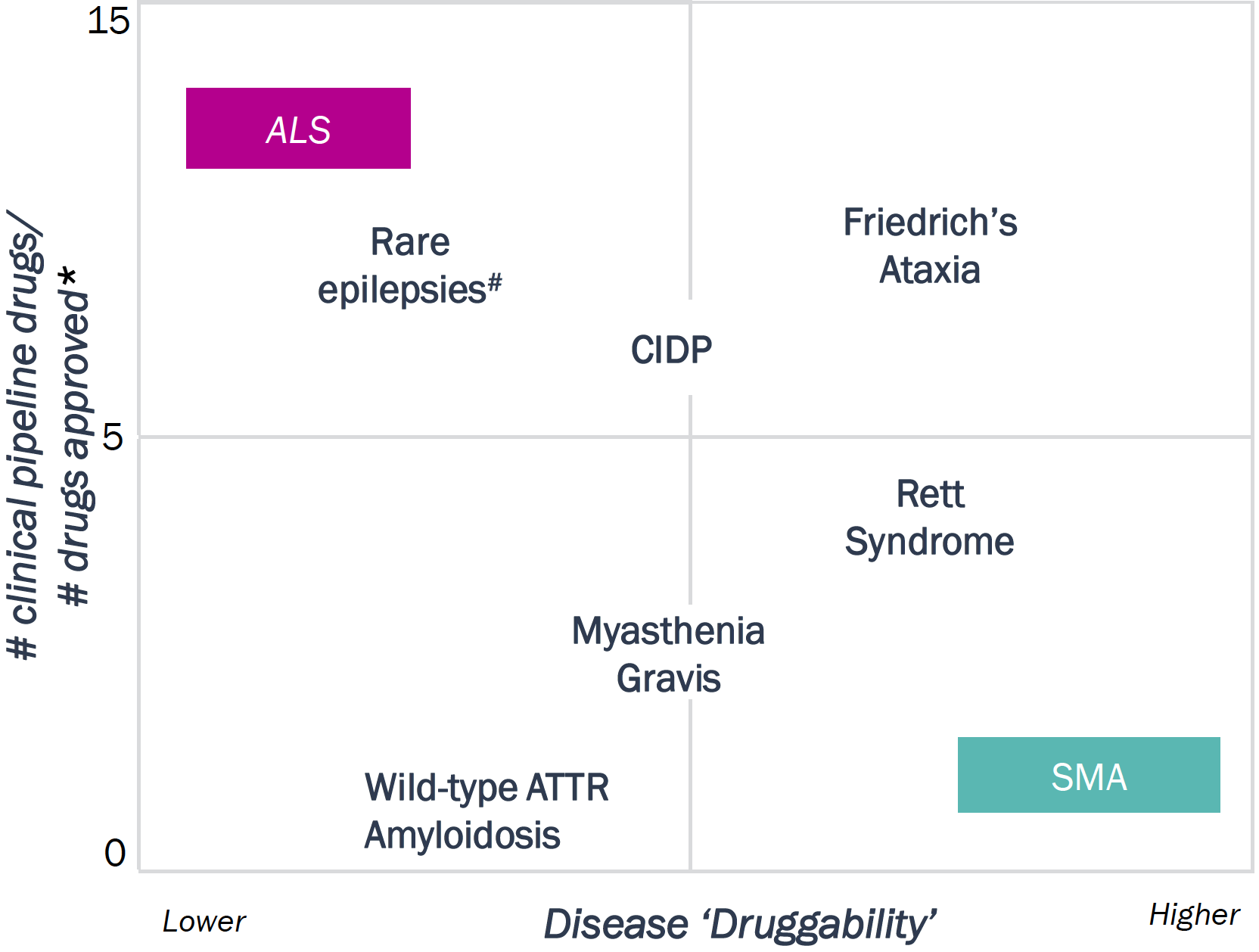
* Counting number of approved therapies in
the last 10 years
#includes Lennox Gastaut, Dravet Syndrome, Tuberous Sclerosis
Legend: ATTR: Transthyretin amyloid cardiomyopathy; CIDP: Chronic Inflammatory Demyelinating Polyneuropathy
| ALS | SMA |
|---|---|
| Disease ‘Druggability’: This qualitative metric reflects 1) the degree to which the underlying etiology is known and 2) heterogeneity in clinical presentation and disease progression | |
|
|
| # of clinical pipeline drugs/#approved drugs*: This ratio is one indicator of the remaining unmet need and level of satisfaction with current therapies. The higher the ratio, the higher the need for better therapies | |
| Ratio = 45:4 | Ratio = 5:3 |
Companies investing in ALS are exploring a diverse set of drug modalities and targets to increase the therapeutic potential relative to current therapies and pipeline competitors
Drug Modality and Target Strategies – ALS

Legend: ASO = Antisense Nucleotide
Clinical development in ALS is aiming to achieve stronger efficacy signals with more validated endpoints; time- and cost-efficient strategies are also being adopted
ALS Clinical Trial Strategies

Legend: : ALSFRS-R = Revised Amyotrophic Lateral Sclerosis Functional Rating Score; Adcomm = Advisory Committee; SPA = Special Protocol Assessment
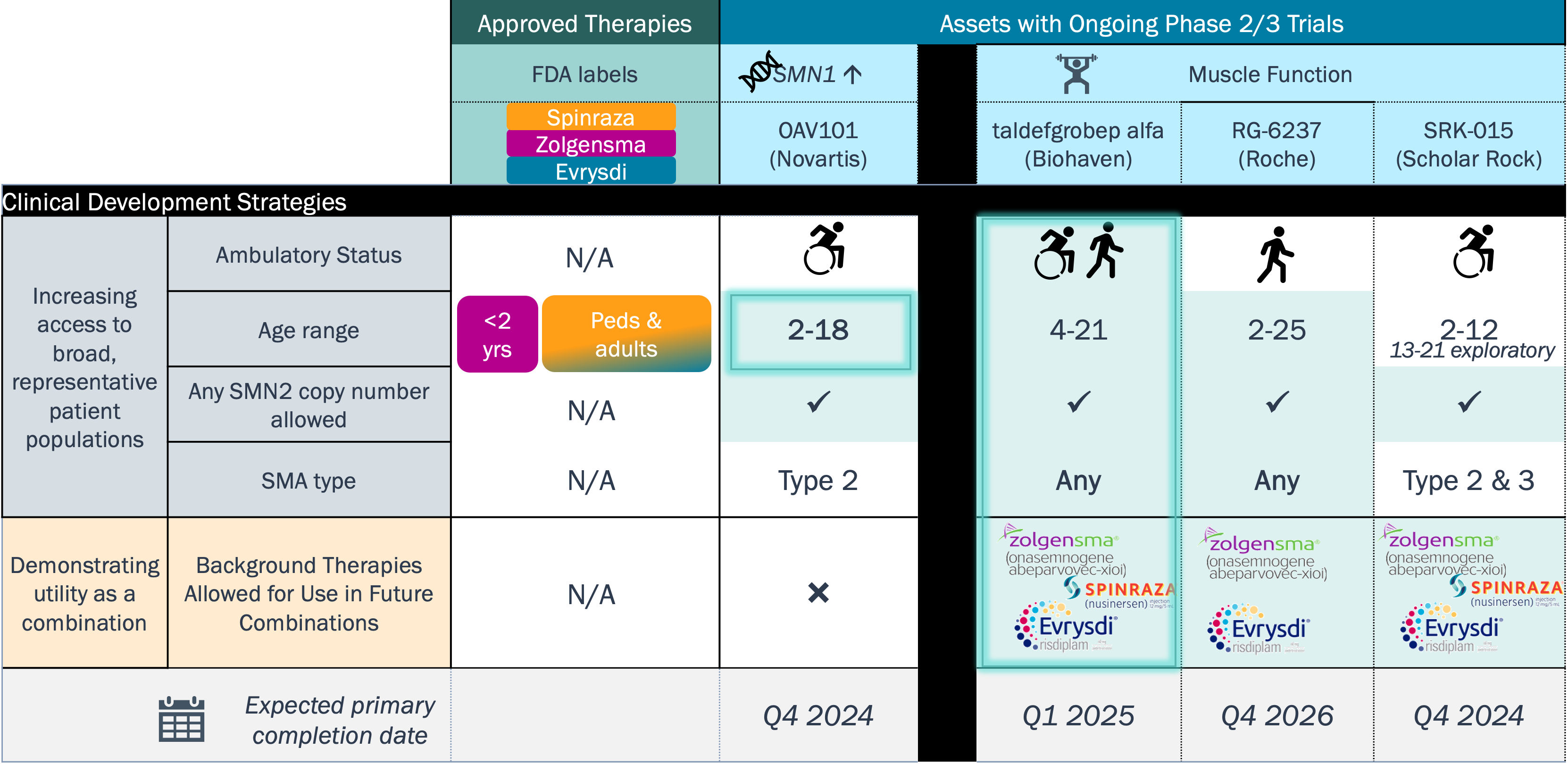
In contrast to ALS, assets in clinical development for SMA aim to augment the effectiveness of marketed products to expand patient eligibility and enhance clinical benefit
Drug Modality and Target Strategies – SMA
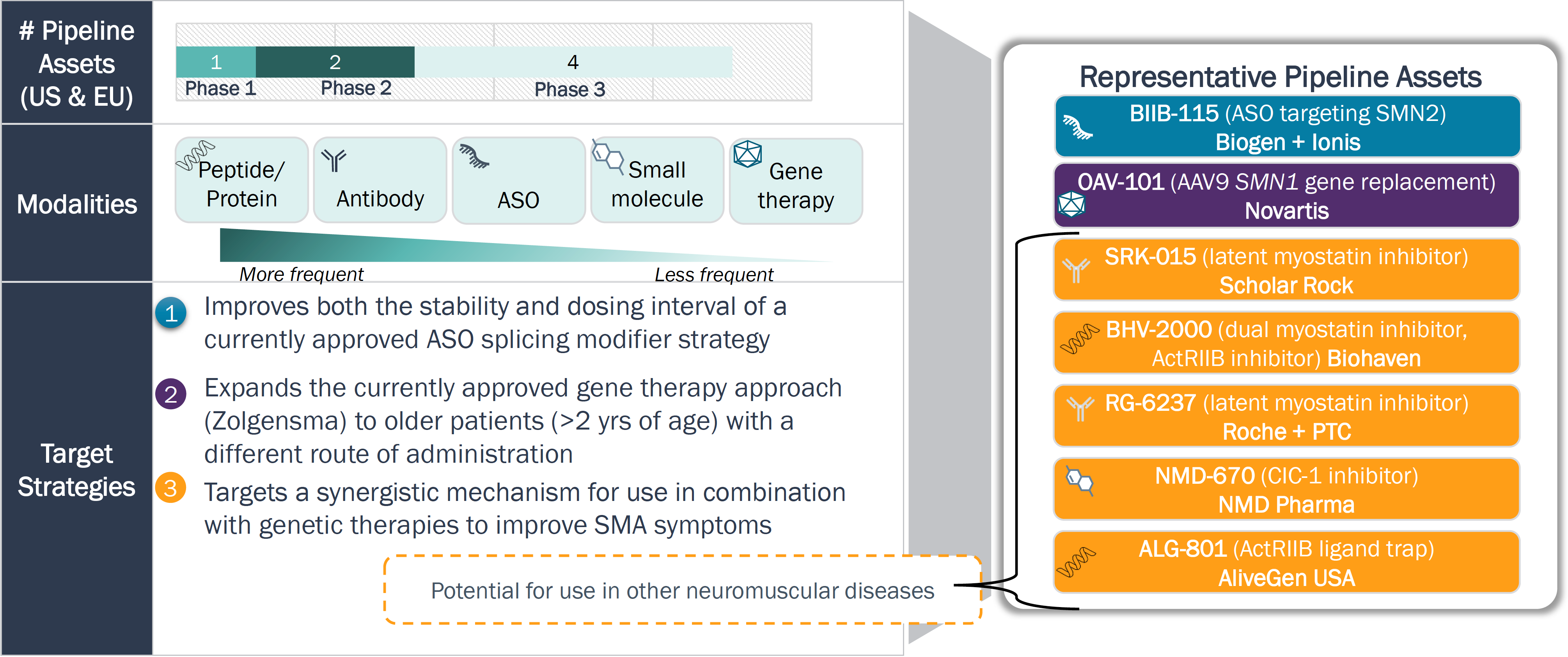
Legend: ASO = Antisense Nucleotide
With regulatory path defined, clinical development in SMA is currently focused on further enhancing the therapeutic benefit in broader patient segments (disease severity and age)
SMA Clinical Trial Strategies
Rare CNS diseases have benefited from an influx of novel therapies, but the remaining unmet needs and clinical development strategies vary by disease and continue to pose a challenge
Outlook on Rare CNS Disease Therapeutics
- There has been an uptick in FDA approvals in the last 10 years for rare
CNS disease, which reflects industry’s increasing investment in these
therapies. This trend can be attributed to the following:
- Multiple clinical development and commercial incentives associated with rare CNS diseases
- Advent of novel technologies (e.g., gene-targeting therapies or therapies that enhance biodistribution)
- Rare CNS diseases vary in their “druggability” and their relative unmet
need, which consequently impacts drug development strategy, as evidenced
by ALS and SMA.
- Several treatments are in development for ALS, reflecting the high unmet need that persists. A variety of approaches and strategic clinical trial designs are being adopted to differentiate from suboptimal standard of care and pipeline competitors.
- Marketed SMA therapies have seen a significant uptake due to NBS, and as a result, fewer therapies are in development than for ALS; drug pipeline in SMA is focusing on maximizing clinical benefit of existing therapies.
- There are two future counter-acting initiatives that could alter the
drug development landscape for rare CNS:
- Inflation Reduction Act (IRA): How will it impact commercial incentives that have been critical for drug innovation, including rare diseases?
- Operation Warp Speed in rare diseases: How will companies alter their drug development strategies in terms of indication selection or investment in cell/gene therapy technologies?
The Authors
Emma Janke,
PhD
Senior
Consultant
|
Pulses are our e-newsletters that look at recent trends and promising directions across life sciences. Add your name to our mail list to receive future updates.